在临床生物样品分析领域,法规的遵循与技术的精准度同等重要。每一次法规的更新,都是行业发展道路上的新标识。
2025年全球生物分析法规持续变革,迎来了多个重要动态,这些动态与现行的ICH M10落地紧密相关。以下将以ICH M10为对照,对全球生物分析法规进行深入剖析。
FDA新规:BMV落幕,M10全面主导
2025年1月,FDA Bioanalytical Method Validation for Biomarkers Guidance for Industry(BMV for Biomarkers,生物标志物生物分析方法验证行业指导原则)发布。
该文件内容不多,比较引人关注的是其对文件发布背景的表述:“2022年11月发布的ICH M10原本预计取代FDA在2018年发布的BMV(Bioanalytical Method Validation)指导原则,但在BMV中包含了化药、生物药以及生物标志物浓度检测所需遵循的方法验证方式,而M10仅将生物标志物置于“相关考虑”部分。随着本指导原则的发布,原有的BMV指导原则将撤销(原文:publication of this guidance, the guidance for industry Bioanalytical Method Validation (May 2018) will be withdrawn)。”
展开剩余86%在此之前,虽然FDA已宣布用于申报的生物分析应遵循 M10,但由于BMV的存在,使得部分要求存在模糊地带。如今BMV退出历史舞台,意味着除生物标志物生物分析外,BMV与M10间的差异点从此统一以M10为准则。同时,鉴于FDA的专业性和权威性,业界在进行生物标志物检测时,除需遵循M10外,也应参考BMV for Biomarkers的意见。
中国药典新动向:9012退场,接轨国际标准
2025年3月25日,国家药监局、国家卫健委发布了关于颁布2025年版《中国药典》的公告。公告附件中给出了药典目录,我们注意到四部目录9000指导原则部分已不见《9012 生物样品定量分析方法验证指导原则》。
由于NMPA早在2023年就发布公告,要求自2023年7月29日起开展的相关研究均适用M10指导原则,各实验室的SOP等质量体系文件应已完成调整。所以,这一举措本身不会对各实验室现行项目的执行产生影响。
在很长一段时间里,9012从国内药品研发和检测的实际情况出发,对规范临床生物样品分析发挥了重要作用。不过9012和M10两者确实存在很多差异,如今9012正式退场,也促使了国内临床生物样品检测机构全面对标国际先进标准,审视现有检测流程和质量体系,进一步提升自身在国际市场的竞争力,更好地服务于全球药品研发。
上述两项内容可视为2022年各主要药监机构支持M10落地后,区域性法规随之进行适应性调整所产生的后续影响。接下来,世界卫生组织(WHO)的文件则是在M10的基础上,针对一些细节作出更为明确的规定。
WHO发布新指导原则:这些细节不容忽视
2025年4月,WHO以第五十八次专家委员会报告的形式发布的生物分析指导原则同样值得关注。该文件的绝大部分内容与M10一致,但在一些细节上具有独特之处。需要注意的是,目前主要法规机构明确遵循的指导原则仍只有M10。不过,WHO的这份文件对M10起到了补充作用,无疑具有参考价值。
对比WHO及M10的区别如下表:
通过以上对比,可清晰看到两份文件之间的差异点,其中部分内容可能需要特殊关注。
1. 对于研究样品浓度持续高于定量上限、需增加更高浓度稳定性样品的情况,建议使用DQC样品
M10中规定,如果研究样品的浓度持续高于校准曲线的ULOQ,则应调高稳定性质控样品的浓度,以反映这些较高浓度样品的稳定性结果。WHO文件中进一步明确建议在此种情况下采用DQC评估稳定性。我们认为这可以引申理解为,用于考察“更高浓度的稳定性”的样品,其浓度不需高于真实样品的最高浓度,采用代表性浓度即可。这为实验室在应对此类复杂样品时提供了清晰的操作指引。
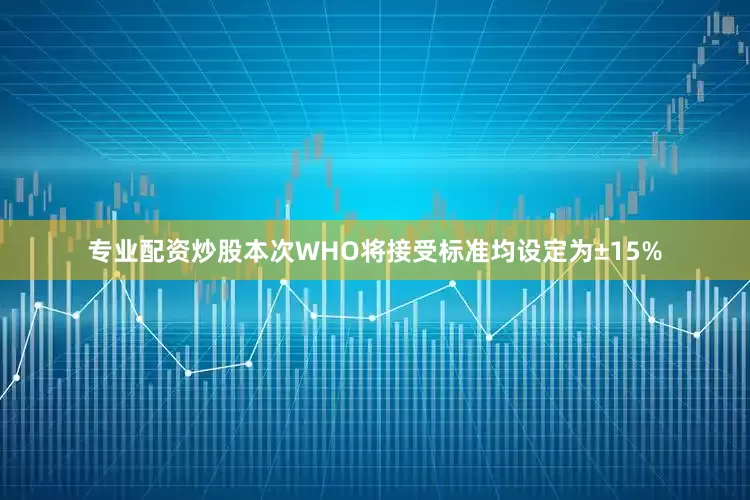
2. 溶液稳定性和全血稳定性的接受标准为±15%
此前M10等指导原则中仅规定了溶液稳定性和全血稳定性的考察方式,但未规定接受标准。本次WHO将接受标准均设定为±15%。
在溶液稳定性方面,由于考察时仅进行简单的稀释操作,且仅做相对性的响应比较,无需定量,因此业内实验室可能会采用更严苛的标准,如±10%。
在临床血浆样品检测中,全血样品的稳定性对检测结果的准确性至关重要。对于全血稳定性接受标准的明确,有助于规范稳定性评估。全血稳定性考察涉及离心操作,且实验结果容易受到全血血浆分配比的影响。在法规未有明确接受标准的情况下,目前部分实验室将接受标准设定为±20%,但在WHO本篇指导原则发布后,这将具有一定的申报风险。
有济实验室始终参照行业高标准进行要求,在溶液稳定性方面遵循±10%的接受标准,在全血稳定性方面则一直遵循±15%的接受标准。
3. 连续ISR失败或应调查
ISR是评估检测结果重现性的重要手段。既往指导原则中大多从整体ISR的角度判断是否存在异常情况,具有一定的滞后性。而在实际操作中,当研究持续时间较长时,大家往往习惯采用滚动ISR的方式,以帮助及时发现问题,确认阶段的检测结果是否准确。若采用滚动ISR的方式,则连续失败可能意味着检测过程存在系统性问题。通过及时调查,实验室能够发现并解决潜在的问题。这一要求对于实操更具指导意义。
这一规则的起源或许可以追溯至GBC(Global Bioanalysis Consortium)在2014年发表的一系列论文。在ICH M10发布之前,GBC曾尝试以行业白皮书的形式推行统一的生物分析执行方式,后来其中的很多意见被M10采纳吸收。在GBC白皮书中关于滚动ISR发生连续失败的叙述如下:
“当采取滚动ISR方式时,如果只分析了少量的ISR样本且结果并不显著异常,则基于单次ISR“失败”而停止分析并启动调查可能并不必要.......应依据科学判断,如果连续两次ISR结果未能满足接受标准,当然应该考虑进行调查。”
“For example, when conducting ISR on a rolling basis, it may not be necessary to halt analysis and initiate an investigation based on the “failure” of a single run if a small number of ISR samples were analyzed and the results were not highly aberrant.......Scientific judgment should be employed and certainly an investigation should be considered if ISR results from two successive runs fail to meet acceptance criteria.”
除上述各点之外,部分内容的执行方式或许仍有待商榷,不过指导原则所指引的方向是具有意义的。
1. 残留评估的新思路
当分析批中出现大量样品超出定量上限(AQL)时,WHO建议使用未稀释的DQC进行残留评估,而非传统的ULOQ样品。这一建议为实验室处理复杂分析批提供了新的思路和方法,有助于更准确地评估残留情况,保障检测结果的可靠性。
但是这在实际操作中存在一定难度。在执行样品分析前,可能难以预计该分析批中AQL样品的数量或比例(例如在设计了不同剂量组的盲态检测中),也就难以判断是否要在特定的分析批中安排未稀释的DQC样品用于评估残留。
2. 关于交叉验证
M10发布之初,交叉验证的评估方式曾引起关注。既往的指导原则参考QC样品的Bia%接受标准或真实样品的ISR接受标准,M10提出了新思路:Bland-Altman作图、Deming回归、一致性相关系数、每种方法得到的研究样品检测浓度-时间曲线作图。
我们在此前关于M10法规的解读(ICH M10: 新法规下生物分析研究思路的探讨)中即指出,这或许表明在交叉验证方面,法规部门似乎开始更关注两组数据之间是否真实存在差异。本次WHO文件延续了M10的态度且更为直接地指出:交叉验证没有明确的接受标准,若观察到偏倚,必须评估其影响,无论偏倚程度大小。
M10发布后,有济临床生物分析团队对于Bland-Altman作图等新颖的评估方式已进行了内部探讨。若采用该方式,需要在进行计算前评估数据是否适用。
3. 关于回收率考察的操作流程
在传统的指导原则中,大多将回收率作为方法验证的重要考察内容。然而,M10首次通过章节目录等形式作出提示,回收率应在MD阶段而非MV阶段进行考察,这在一定程度上弱化了回收率的地位。WHO也进一步指出回收率应在MD阶段进行,尽管它同时要求了应提交相关数据以支持MV,但这也明确了回收率在方法开发和验证过程中的操作流程。
但问题是方法开发和方法验证的法规依从性不同。方法开发阶段往往不需保存完整记录和稽查轨迹,数据可不经QA审查。那么如何以MD的数据提交符合申报要求的实验结果?这可能是需要进一步探讨的话题。
有济医药:紧跟变化、专业合规
在生物分析法规不断更新的当下,有济医药始终保持敏锐的洞察力,以积极的姿态紧跟法规动态。我们严格遵循 ICH M10 以及各国最新的法规要求,凭借先进的检测技术,为临床生物样品检测提供精准、可靠的服务。无论是在法规遵循还是技术能力上,有济医药都展现出了专业的态度和卓越的能力,持续为临床药代动力学研究提供高质量的数据支持,助力新药研发加速前行。
参考文献:
[1]ICH M10 生物分析方法验证及样品分析, 2022
[2]Bioanalytical Method Validation Guidance for Industry, FDA, 2018
[3]Bioanalytical Method Validation for Biomarkers Guidance for Industry, FDA, 2018
[4]9012 生物分析方法验证指导原则, 中国药典2015版
[5]Annex 6 Guideline on bioanalytical method validation and study sample analysis, WHO, 2025
[6]Repeat Analysis and Incurred Sample Reanalysis: Recommendation for Best Practices and Harmonization from the Global Bioanalysis Consortium Harmonization Team [J], The AAPS Journal, Vol. 16, No. 6, November 2014
[7]Small Molecule Specific Run Acceptance, Specific Assay Operation, and Chromatographic Run Quality Assessment: Recommendation for Best Practices and Harmonization from the Global Bioanalysis Consortium Harmonization Teams [J], The AAPS Journal, Vol. 16, No. 5, September 2014
发布于:天津市中国股票配资网上提示:文章来自网络,不代表本站观点。